
For a full list of BASHing data blog posts see the index page. ![]()
Combinations from 2 lists: speed trials
This post was inspired by a recently published scientific paper describing how Python was used to build a list of a million scientific names. Each name was composed of parts taken from a list, and combinations of those parts were generated.
The result was something like a Cartesian product, about which I've blogged before. This time I was interested in performance: how does the time required to get a result vary with the number of combinations to be built?
My raw material was a shuffled list of 497113 unique genus names, "gen", and a shuffled list of 806105 unique species names, "spe". What I wanted to build were scientific names of species, like these examples:
Passer domesticus
#a real species, the house sparrow
Eucalyptus domesticus
#a non-existent species of tree
There would be mismatch cases, for example a feminine-gender genus paired with a masculine-ending species, but this is just a combinatorial exercise, not a proper name-building one.
I tried two different combination-building methods. The first uses nested BASH loops:
for i in $(<gen); do for j in $(<spe); do printf "%s\n" "$i $j"; done; done
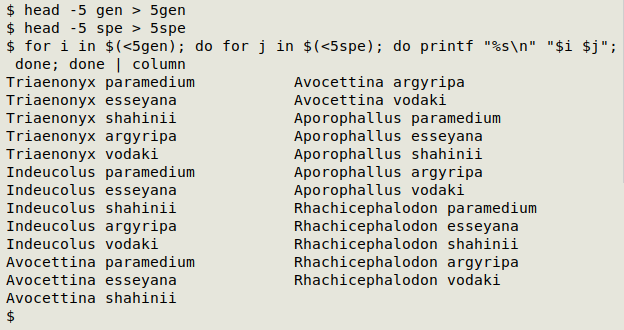
The second method puts the species names in an AWK array, then builds the combinations from genus names and array elements:
awk 'FNR==NR {a[$1]=$1; next} {for (i in a) print $0,a[i]}' spe gen

The species order of the AWK result is different from that in the BASH result because elements in AWK arrays aren't ordered by default. The AWK and BASH results are identical if both outputs are sorted.
I used head on the shuffled lists to build files with 10, 20, 50, 100, 200, 500, 1000, 2000, 5000 and 10000 genus or species names. In the first pair of trials I compared BASH and AWK speeds in building genus-species combinations with a constant 10 genus names, sending the combinations to /dev/null. In the second set of trials the combinations were built from a constant 10 species names, with increasing numbers of genus names. The results are shown below as log-log plots.
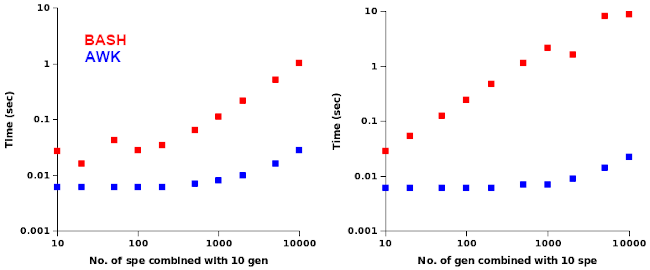
The AWK command was considerably faster and the AWK time increased more slowly with more names to be combined than did the BASH time.
The "jiggles" in the BASH plots were reproducible, and I'm not sure what was happening there — maybe a change in memory use? It's also curious that an increasing load on the outer BASH loop slowed the process more than an increasing load on the inner loop.
In the third trial I compared BASH and AWK speeds in combining equal numbers of genus and species names. As shown on the log-log plot below, the AWK command was again the race winner and settled down (at higher numbers of names) to being about 60 times faster than the nested BASH loops. In other words, it would take BASH 10 minutes to do what AWK did in 10 seconds.
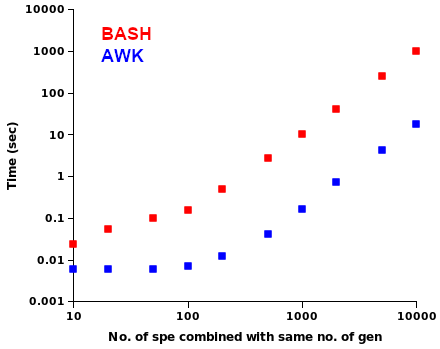
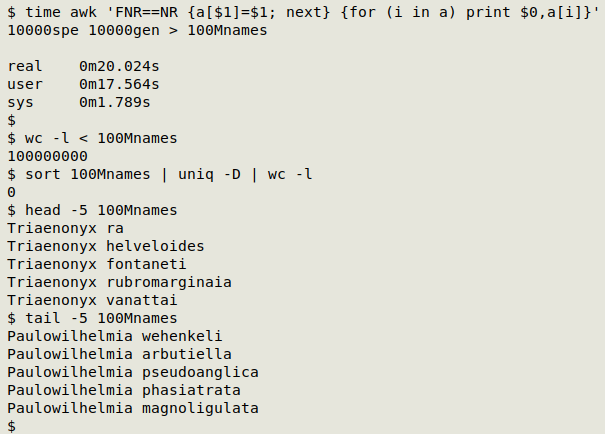
More on that last AWK combination below. Writing 100 million unique genus-species combinations to a 2.3GB text file only required about 20 seconds on my desktop (6 core Intel Core i5-9500T, 8GB RAM).
Last update: 2021-12-01
The blog posts on this website are licensed under a
Creative Commons Attribution-NonCommercial 4.0 International License