
For a full list of BASHing data blog posts see the index page. ![]()
Python and shell tools
I'm not a pythonista, and what little I know about Python for data work amounts to a few published recipes. Out of curiosity, I sometimes re-do those recipes with the GNU/Linux tools I use every day. Below are three such re-doings from Python 2.7 (default on my Debian 10 system, but soon to reach end-of-life).
Please note that this post isn't meant to be a "which is best?" contest between Python and shell tools. Each world of commands has its pro's and con's, and Python users have access to a large number of general and specialised data-processing tools. Personally, I like the versatility of shell tools and command chains, and I like AWK's speed and flexible syntax (as readers of this blog will know).
If there's a data processing task you do regularly with Python and you're wondering what the shell-tools equivalent might be, email me about it and I'll see what I can come up with.
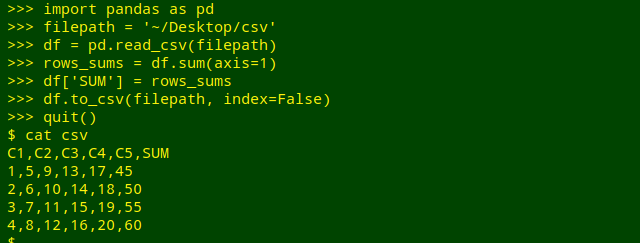
Add row sums to a CSV. This example comes from the Rosetta code website and uses the pandas library. The demo file on my desktop is here called "csv" and the Python commands do an in-place file replacement:
C1,C2,C3,C4,C5
1,5,9,13,17
2,6,10,14,18
3,7,11,15,19
4,8,12,16,20
Being strongly AWK-ophilic I would probably do the row sums with GNU AWK 4 and its -i inplace option:
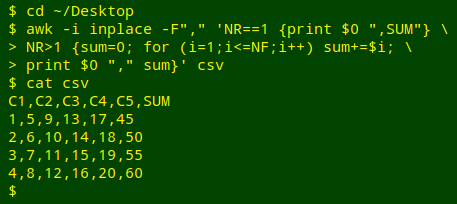
AWK is told with -F"," that the field separator in "csv" is a comma. To the first line (NR==1) AWK adds a comma and the word "SUM" as field name (print $0",SUM"). In succeeding lines (NR>1) the variable "sum" is first set to zero (clearing it for use on that line). AWK then works through the line field by field (for (i=1;i<=NF;i++)) and increments "sum" with the value in the field (sum+=$i). At the end of the line, AWK prints the existing line followed by a comma and the value of "sum" for that line (print $0","sum).
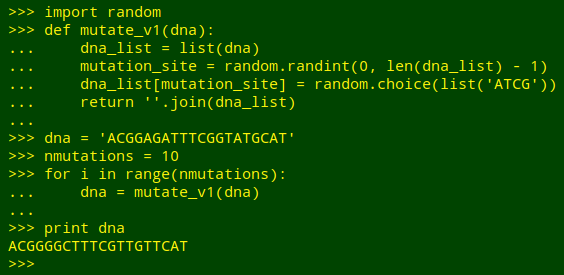
Mutate some DNA. I found this task in Illustrating Python via Bioinformatics Examples, a very clearly written website by Hans Petter Langtangen and Geir Kjetil Sandve. The goal is to randomly replace the As, Ts, Gs and Cs in a DNA sequence with A, T, G or C. The authors write "A function for replacing the letter in a randomly selected position (index) by a random letter among A, C, G, and T is most straightforwardly implemented by converting the DNA string to a list of letters, since changing a character in a Python string is impossible without constructing a new string. However, an element in a list can be changed in-place":
ACGGAGATTTCGGTATGCAT #sequence to be mutated, in this case 10 times
I've seen a fairly complicated AWK script on the Web to mutate a DNA sequence, but the job can also be done with simpler shell tools. Two things need to be (more or less) randomised: the position in the sequence where an A, T, G or C substitution will happen, and the choice of A, T, G or C to replace the original. To do the first, I can pick a random number from 1 to the length of the sequence using shuf:
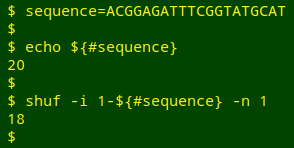
${#sequence} gives the length of the "sequence" string. shuf's -i option allows you to choose a range of numbers to be shuffled, in this case from 1 to the length of the sequence. The -n 1 option selects the first of the shuffled numbers.
To select the A, T, G or C as a replacement:
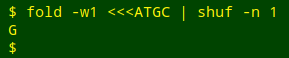
fold -w1 puts the 4 letters in the string "ATGC" into a vertical list. shuf -n 1 shuffles the list and returns the first letter.
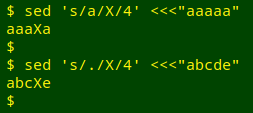
The "mutation" substitution can be done with sed, which allows for replacement of the "nth" occurrence of a searched-for pattern. For example, if I want to replace "a" with "X" at the 4th occurrence of "a" in "aaaaa", I write sed 's/a/X/4' <<<"aaaaa", and if I want to replace the 4th character (of any kind) with an "X" in a string of characters, I write sed 's/./X/4':
In the mutation command, the "X" will be $(fold -w1 <<<ATGC | shuf -n 1) and the position of the mutation will be $(shuf -i 1-${#sequence} -n 1). To allow the shell to interpret those expressions, I'll need to change single quotes to double quotes in the sed command:
sed "s/./$(fold -w1 <<<ATGC | shuf -n 1)/$(shuf -i 1-${#sequence} -n 1)" \
<<< "$sequence"
To test this command I'll run it 10 times separately on the same sequence, each time highlighting the replacement A, T, G or C. Notice that sometimes the replacement is the same as the original:
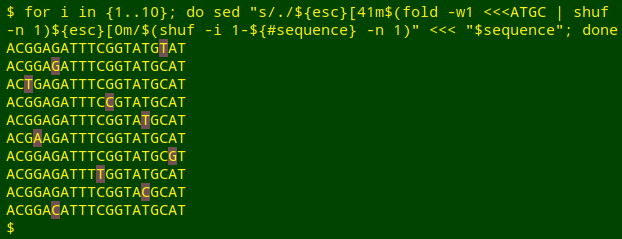
Before running this command I defined the variable "esc" as $(printf '\033'). For more on colorising characters, see this earlier BASHing data post.
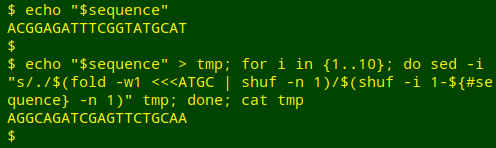
Finally, to "re-do" the Python commands I'll mutate the sequence 10 times progressively by using sed's "-i" (in-place) option. Because sed only does in-place replacements on files, not strings, I'll first save the sequence as the temporary file "tmp". After 10 mutations of "tmp", I'll see what it looks like with cat:
echo "$sequence" > tmp; for i in {1..10}; do sed -i "s/./$(fold -w1 <<<ATGC | shuf -n 1)/$(shuf -i 1-${#sequence} -n 1)" tmp; done; cat tmp
According to Bruce Barnett, the sed s/./replacement/n trick will only work up to n=512. I'm running GNU sed 4.4, and it can do replacements up to at least n=1,000,000. (I haven't tried longer strings.)
Get a field tally. The demo file (again on my desktop) is "records", which is a TSV with 42,637 lines plus a header line, and 132 fields. I'll import the file into the pandas environment and get some info on the file:
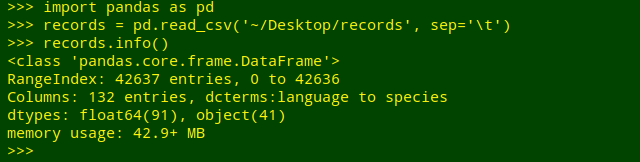
What I'd like is a tally of the unique entries, including blanks, in field 61 of "records", which has the field name "waterBody". Python does this very neatly, though I have to tell it to sort by the tallied items (sort_index()) and to include any blank entries (value_counts(dropna=False)):
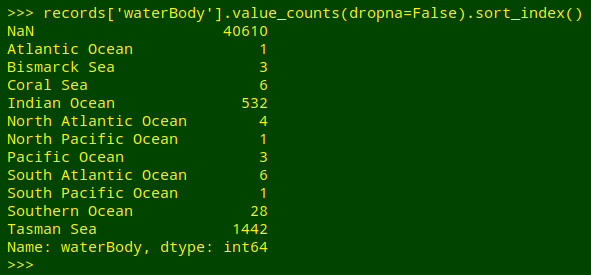
In the shell I can get a similar result in several ways. One method would be to trim off the header line with tail, cut out field 61, sort the field and uniquify it with counts:
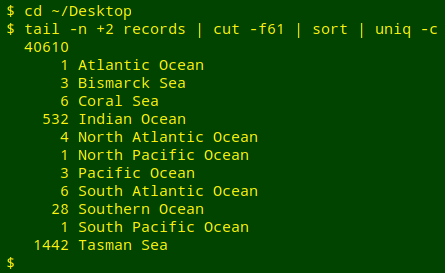
I could also use GNU datamash, then prettify the results with AWK:
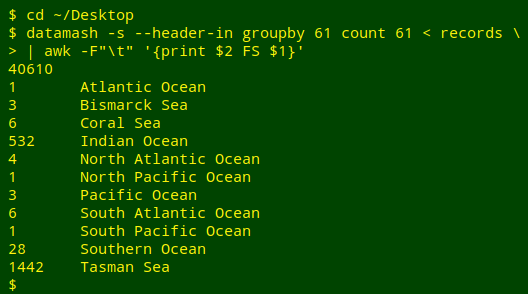
A third method would be to put counts of the unique entries in field 61 into an AWK array, then iterate the array values by index value and sort the results by index:
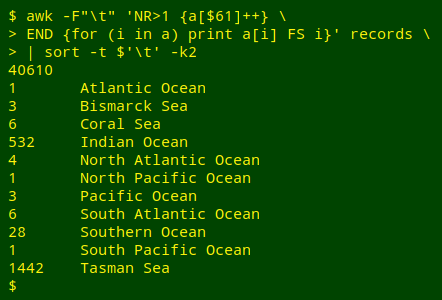
For an introduction to AWK arrays, see the 4-part series beginning here.
Last update: 2019-11-22
The blog posts on this website are licensed under a
Creative Commons Attribution-NonCommercial 4.0 International License